Requirements
VERSION 3.0
Team Yaxonomic
Evan Bolyen
Mike Deberg
Andrew Hodel
Hayden Westbrook
Faculty Mentor/Project Sponsor: Viacheslav “Slava” Fofanov, PhD
3. Problem and Solution Statement
5. Non-functional Requirements
Artifact:
A collection of output data from a single step of the pipeline.
Artifact Key:
An opaque handle to an artifact. How YAX tracks artifacts.
Bowtie2:
Tool for the alignment of genetic reads to reference sequences.
Coverage:
A measurement of how much of a reference sequence is covered by reads.
FASTA file:
File format used for storing genetic sequences with header and sequence
Genbank:
Repository of genetic sequences.
Genetic sequence:
A string of characters representing nucleotides that is the output of a sequencing machine. The characters used to represent nucleotides are A,T,C and, G in some cases N and other characters will be used as placeholders for unknown nucleotides. This is specifically the IUPAC alphabet.
GI:
A unique numeric identifier assigned to every genetic sequence observed by NCBI. Every genetic sequence observed will have a unique GI, even if they are from the same species or even the same individual.
High-throughput Sequencing:
A term describing modern sequencing techniques
Hit:
When a read is mapped to a TaxID
In List:
Metagenome:
The presumed set of all DNA in a discrete environment. (The function capacity of that environment)
NCBI:
National Center for Biotechnology Information
Nucleotide:
A single nucleic acid of which DNA is composed of.
Pathogen:
A harmful biological agent.
Read:
A genetic sequence from an unknown species that comes from a sample from the environment.
Read Fragment:
A read that has been processed by ReadPrep
Reference:
A genetic sequence contained within the NCBI database that belongs to a known species.
RunID:
Identification of a single complete branch of a YAX run. Used to manage branch data; represents an amalgamation of Artifact Keys in the pipeline.
Sample:
Taxon:
A particular node in the taxonomy tree.
Taxonomic Assignment:
The pairing of a sequence to a taxonomy
TaxID:
A unique numeric identifier for a node in the taxonomic tree of NCBI’s database.
TaxID Tree:
File containing a set of TaxIDs and metadata that can be used to build a tree representation of taxonomic data
TaxID Reference Set:
FASTA file that contains the set of sequences for TaxID references
TaxIDBranch:
Tool for isolating branches of taxonomy.
Wall-time:
The quantity of time a process executed.
YAX:
A working name for the taxonomy assignment pipeline. We do not have an official name yet, but we recommend YAX (YAX assigns taxonomy).
This requirements document is intended to familiarize the reader with the tasks that the taxonomic assignment pipeline (hereby referred to as YAX) will be expected to accomplish. Any basic computer background should be sufficient to understand the requirements. Some minor bioinformatic knowledge may be required which can be gained from any source discussing the topic of read to reference genome alignment.
The primary function of YAX will be to manage the lengthy process of identifying genetic sequences collected from the environment. It is expected that the computational timeframe of this process will be fairly lengthy and that the capacity to recover from an error at some point in the process will be of paramount importance. The intention of this is to eliminate the need to restart the entire process from the beginning in the event of such a failure. In this way as little time as possible will be lost in what will already be a time consuming process.
This state aware system will also be conducive to any user need of rerunning a portion of YAX. Since YAX with have knowledge of the various states it has already created it should be a relatively trivial matter to append to those states additional pieces of data. For example the addition of reads to an existing run. This system will be entirely decoupled from the modules of YAX.
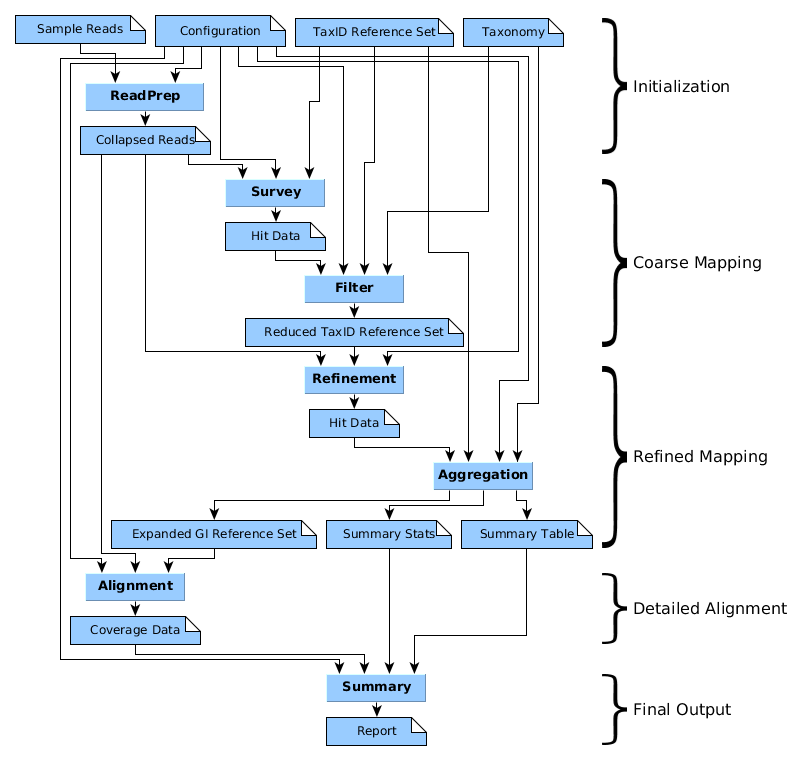
The modules of YAX will include readprep, survey/refinement, filter, aggregation, alignment and, summary. Readprep will work with the initial read input from the user. It will evaluate them based on quality and length, is will chunk reads if necessary and finally collapse any duplicate reads into sets that will be used throughout the other modules. Readprep will record some basic information that will be necessary for the useful output of YAX.
Survey and Refinement are actually the same module, the only difference being that Survey will work on all references found in the National Center for Biotechnology Information’s (NCBI) taxonomy database while Refinement will work on a reduced set of references produced by the Filter module based on hits found in Survey. Filter specifically receives data from Survey in the form of taxonomy identifiers (TaxIDs). These TaxIDs are used to find reference sequences that Refinement will use to build new Bowtie2 index files used for alignment to a reduced set of references.
Aggregation is fundamentally the same module as filter and will be used again after the Refinement module. Aggregation will take the output of Refinement which is again in the form of TaxIDs. These will be filtered once again based on various user inputs and finally a set of associated Genbank Identifiers (GIs) are compiled based on the decided set of TaxIDs. This output is the primary difference between Aggregation and Filter as Aggregation outputs a set of GIs while Filter outputs a set of TaxIDs.The TaxIDBranch tool, developed separately by Fofanov Labs, will be responsible for identifying these associated GIs and building a FASTA file which will be used in Alignment.
Alignment will utilize the tool Bowtie2 to do an alignment of the initially provided reads to the GIs found in Aggregation. The intention is that the Bowtie2 call will allow the user to utilize the full range of functionality and will produce information used to summarize the coverage of reads on references. Finally Summary will summarize this data in a single-document report.
This modular design is intended to extend the significance of YAX as much as possible by allowing the modules to be replaced as new technologies are developed with as little rework as possible. It will also aid in the development of YAX itself to allow an iterative approach to the implementation of the entire system.
The project’s sponsor, Dr. Viacheslav Fofanov, is an assistant professor in the informatics and computing program at NAU. His research focuses on detecting pathogens in environmental samples by utilizing high-throughput sequencing data. This research is important because it provides a line of defense against the spread of dangerous pathogens.
To accomplish this, rapid assessment of the samples is necessary. Using traditional selective or differential media to culture organisms can be especially time-consuming. Worse, not all (in fact most) pathogenic organisms cannot be cultured in a lab.
A relatively modern alternative to this problem is high-throughput sequencing, which offers the ability to quickly collect large volumes of data about the DNA present in a sample. There are two general approaches to this: amplicon and shotgun sequencing. Amplicon sequencing chemically targets known areas of a genome, while shotgun sequencing pulls out random fragments. Because the goal is to collect community information spanning bacteria, eukaryotes, and viruses, targeting any one area of a genome will not work well as these groups are too different. That leaves shotgun sequencing, which will produce information about the entire metagenome, but at the cost of not knowing where any particular sequence came from in a genome or even the genome it originated from. Furthermore these reads are generally short, spanning a couple-hundred nucleotides.
NCBI maintains databases that contain information on organisms which it has an assigned TaxID and a list of GIs. Most importantly for our purposes it maintains associations linking the two, so given an organism one can retrieve a TaxID and all the GIs associated with it. However, these databases are massive, and it is unlikely that a given read will ever match a reference exactly. This means efficient fuzzy fragment matching of these massive datasets is needed. Additionally, as all life (with the potential exception of viruses) has a common origin, many components of a genome are shared. This complicates the search as the detection of a match does not mean it is conclusive evidence for the presence of a given taxon.
What is needed is a system that can: rapidly match metagenomic shotgun reads to sub-sequences of a reference database; identify which reads are informative and which are not; draw conclusions on community membership; and provide some quantitative measure of confidence in the those conclusions.
YAX will be a pipeline that should determine what species are represented within a sample of genetic data. The pipeline will use two primary sources of data. The first are the reads that are the product of sequencing environmental samples. The second are the references that come from the NCBI database. YAX will also utilize a configuration file that the user can edit to manipulate different settings. The reads will be mapped to references to determine what species are represented in the sample. The output of the pipeline will be a number of PDF documents that will describe and visualize the results.
The actual read preparation, aligning of genetic sequences, and mapping of reads to references will be taken care of by tools that are already implemented. Because of this, the system to be developed will primarily be responsible for moving data between separate modules of the pipeline. As a result, one of the core functionalities of the pipeline is a state system that will keep track of what step of the pipeline the execution is currently in, and that will allow the user to return to any previous state and branch execution into another run of the pipeline using different data or configuration options.
Finding the identity of an unknown sample sequence is time consuming and requires the management of extremely large data sets. For this reason, it is expected that coarse decisions are made very rapidly based on the total dataset to shrink it into manageable sizes for more comprehensive analysis. In order to achieve the highest level of accuracy and precision while successfully managing the large data sets presented in this problem an iterative filtering process will be used.
This means that the same general process will be repeated multiple times as it reduces and fine tunes the parameters during each iteration.
The run time of the YAX is expected to be a relatively long period of time, up to a number of weeks. Because of this, error recovery is a major issue to be addressed by the implementation of the YAX. This must be done in a fashion that relies on as little recomputation as possible. This means that the various steps and phases of the project must report accurately on their successful completion or error state and must also store the data in such a way that the damaged or incomplete section is the only part that needs recomputation.
Figure 1. An outline of the module associations with corresponding artifact inputs and output. It should be noted that each module and artifact is a requirement given for implementing the Taxonomy Assignment algorithm.
Ultimately the greatest risk is present in the final output of YAX. If this information is inaccurate, then incorrect or erroneous conclusions could be drawn. These conclusions could go on to impact a vast array of situations from public health to personal reputation. The most likely risk would be the identification of an organism that is not present in the samples. This kind of false positive could significantly alter any kind of published results. The worst case of this scenario could cause serious implications for the researchers' careers, as they would lose credibility and may have future funding opportunities adversely impacted. Any published works based on such erroneous data may also be subjected to retraction.
Alternatively the most severe risk would be a false negative, where a species that is represented within the samples is not identified by YAX. If YAX failed to identify the presence of a potentially harmful organism, there could be serious public health consequences. This situation however, is unlikely to occur, and is even less likely to cause serious problems as YAX is not meant to be relied on for pathogen detection. Either way, the most fundamental way to avoid both false positives and false negatives is to use YAX as one of many identification sources in all critical health-related investigations.
Many researchers do not have access to local superclusters and may rent CPU-hours from cloud based services such as AWS or RackSpace. Additionally if institutional machines are available, there is still the opportunity cost associated with dedicating CPU-hours to any given task. As a result, it is important that YAX is effective in its use of wall-time. Should a runtime-failure occur, it would be disastrous if the entire process needed to be reconstructed from scratch. Failing to create an effective system to “checkpoint” the current state would result in wasted dollars and/or opportunity when such a failure occurs. Most commonly we expect this to be the process running into hard-limits on wall-time, which may be measured in weeks. Recomputing weeks of effort because the walltime was off by a day or two is simply not admissible.
The impact on human resources that are potentially standing by is also not insignificant. YAX is very likely a single step in a chain of investigation. If it does not run in the most efficient manner possible, with accurate run-time failure recovery, resources that are waiting on its finding down the line could be blocked from progression. Obviously this could have a snowballing effect particularly if some step of a process, time in a sequencer for example, needs to be accurately scheduled ahead of time and could result in a significant monetary wastes and missed opportunity.
After the completion of this semester we will have a final requirements document in hand. Based on this sets of test data will be created to provide smaller sample sets of data that can be used to quickly debug the software. The typical amount of data YAX will handle is much too large for initial testing purposes, so the creation of a smaller test data set is crucial.
Implementation will begin with the prototyping of the state system in general so that it can be used throughout implementation of the other modules. Because the state system is relevant through every step of the pipeline, further iteration on the state system will continue as modules are added to the pipeline. Implementation will continue in the order of modules in the pipeline. With the exception of the Survey and Refinement modules which are the same module with the same inputs but of slightly reduced size. The first time the module is given the full precomputed taxonomy tree and TaxID reference set (Survey) and the second time with the reduced TaxID reference set (Refinement). This module will be developed once prior the Filter module and then placed in the workflow again after the Filter module. Similarly the steps of Filter and Aggregation are the same module except for output. Refinement of the reference set is required in either case.
Additionally, the conda packages will be developed alongside to make continuous integration and local tests of the system possible. This will additionally assure that the system can be rolled out on various platforms without issue.
Figure 2. Gantt chart of project timeline.